Antibody prediction software. List of protein structure prediction software 2019-02-05
Is there any tool to predict antigenicity

There are two disadvantages: first, the Kabat scheme is so widely used that some confusion can arise; second, Chothia et al. Antibody informatics tools can help improve our understanding of immune responses to disease and aid in the design and engineering of therapeutic molecules. The range varies from 0 to 1, with a higher score indicating a better structural match. The output from EpiPred is a ranked list of the surface patches on the antigen that could form the epitope. An efficient computational method to predict antibody-specific epitopes at the residue level based on neutralization panels of diverse viral strains. Antibody engineering is a promising field for the treatment of diseases, including cancer, and inflammation.
SAbPred: a structure

Also, the leader sequence should not be included in the sequence selected for antibody generation. The tool uses a proprietary Antibody Name Dictionary model - a collection of protein names, synonyms, and symbols recognized as antibody names which are verified through a full-text search of scientific publications in HighWire Press. Models of the antibody Fv can be used in many other ways including paratope prediction , , epitope prediction , and protein docking. The higher the i-Patch score the higher the likelihood that the residue is in contact with the antigen and is part of the paratope. Continuous epitopes consists of a contiguous sequence of amino acids in a protein. A user may export the top N ranked paratope residues and annotate them with a chosen numbering scheme.
SVMTriP: a tool to predict linear antigenic epitopes

A user may toggle each motif on and off and filter by those that are exposed to the solvent. The output from EpiPred is a ranked list of the surface patches on the antigen that could form the epitope. Cyrus customers include 10 of the top 20 Global Pharmaceutical firms and is financed by leading investors in both Technology and Biotechnology, including Trinity Ventures, Orbimed, Springrock Ventures, Alexandria Venture Investments, and W Fund. The program builds a model from the amino-acid sequence and calculates an estimated accuracy for segments of the model. Examining the conformations of the loops shows that L30 is the correct position. Jeffrey Gray at Johns Hopkins University, but those methods have remained difficult to access.
Cyrus Biotechnology announce antibody structure prediction software
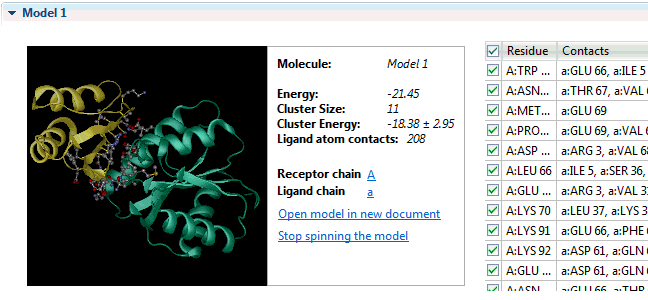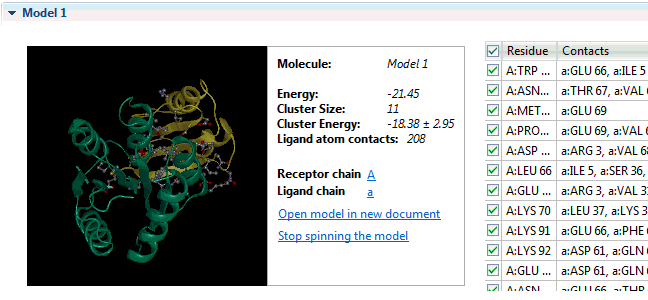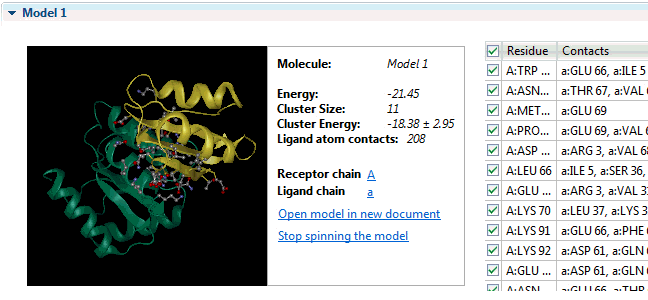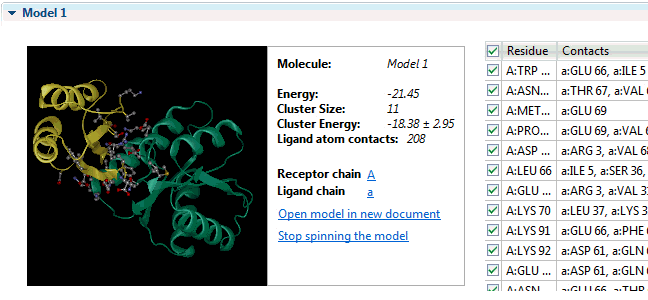
Currently, the majority of available epitope prediction methods focus on continuous epitopes due to the convenience of the investigation in which the amino acid sequence of a protein is taken as the input. An overview of each algorithm is given in the following sections. Hydrophilicity plots as described by assign an average hydrophilicity value for each residue in the sequence and the highest point of average hydrophilicity for a series of contiguous residues is usually at or near an antigenic determinant. This enables scientist using the software to work on a wider variety of common monoclonal antibody engineering problems. Surface region or regions of high accessibility often border helical or extended secondary structure regions.
Cyrus Biotechnology Announces Release of Antibody Structure Prediction Software for Biologics Drug Development and Discovery

It can provide insight into the development of escape mutations. The method primarily utilizes neutralization potency data over a set of diverse viral strains representing the antigen, and enhanced accuracy is achieved by incorporating information from an unbound structure of the antigen. The table presents the chain name, residue number N. The antibody structure and sequence are coloured according to the i-Patch score warmer colours indicate a higher score and confidence that the residue will be part of the paratope. The data are available by clicking. Sequence numbering The sequence numbering application Figure may be used to annotate either a single or multiple antibody variable domain amino-acid sequences. Models of the antibody Fv can be used in many other ways including paratope prediction , , epitope prediction , and protein docking.
Antibody H3 Structure Prediction
Fv structure modelling and annotation The antibody Fv modelling application Figure accepts as input the heavy and light chain amino acid sequences of the molecule. Epitope prediction: EpiPred Residues that the antibody interacts with on an antigen form the epitope. Allows to predict antibody—antigen affinity changes upon mutation which relies on graph-based signatures. Users can customize structural quality and clustering parameters e. What do you mean by antibody production? NovaCloud Take advantage of our cloud computing resources by running protein predictions though our , or run predictions locally on a Mac or Linux computer. If you mean peptides that acts against antigen, then you can mention it as antimicrobial peptide.
SAbPred: a structure

Peptide sequences with length of 10-20 residues minimize synthesis problems since they are reasonably soluble in aqueous solution and may have some degree of secondary structure. Note That in their latest paper Al-Lazikani et al. In this scheme, the locations of deleted residues are indicated with. Cyrus Bench ® is based on the Rosetta software from Prof. A user may export the top N ranked paratope residues and annotate them with a chosen numbering scheme.
Antigen Prediction Tool
In the absence of an experimentally determined structure, a toolbox of computational methods are required to predict such features. Second, the numbering adopts a rigid specification. Structure determination of antibody—antigen complexes can provide epitope information at the atomic level, but in many instances, atomic-level complex structures can be challenging to obtain. A user may toggle each motif on and off and filter by those that are exposed to the solvent. Structural insights gained through modelling also allow potential issues with in vitro development to be identified and overcome.
Antibody H3 Structure Prediction

In: Antibody Engineering Lab Manual Ed. The combined method predicts the final score that combines the predictions from 7-allele method and immunogenicity method. Some of these antibodies will target the linker region and the carrier protein itself in addition to the peptide of interest. When used with NovaDock, the software allows you to predict the structure of antibody-antigen complexes. For additional information including pricing, please.
orgmode.com : Prof. Andrew C.R. Martin's Group at UCL

As output, each residue is annotated with a score. For single sequences a user should paste the raw sequence e. Discontinuous epitopes consist of sequences of amino acids that are not contiguous but are brought together when the peptide chain folds or by the juxtaposition of two separate polypeptide chains. The NovaFold Antibody algorithm utilizes a combination of homology modeling and ab initio loop prediction, resulting in highly accurate predictions. In addition, information on the availability of an individual hybridoma and its Mab product are included. A list of residues is returned and epitope patches may be visualized on the structure. Antibody structures have the potential to be useful during drug development, allowing the implementation of rational design procedures.